郭帆/王红梅团队解析小鼠早期胚胎和生殖细胞发育过程中DNA羟甲基化概观及其调控
长期以来,DNA甲基化(5mC)作为哺乳动物中最为经典的DNA共价修饰被广泛研究,其对基因表达调控、转座子沉默、基因印记以及X染色体失活等生命过程至关重要。直到2009年,Anjana Rao实验室在Science首次报道了在哺乳动物细胞中,存在由一类DNA双加氧酶Tet1/2/3介导的基于5mC氧化而产生的新的DNA共价修饰——DNA羟甲基化(5hmC)1;随后,关于5hmC的生物学功能及其意义成为表观遗传学领域有待回答的重大科学问题之一。经过领域内十余年的研究,目前关于5hmC的生物学功能主要存在两种观点:一是5hmC会介导DNA的主动去甲基化,从而产生新的未经修饰的胞嘧啶。体外支持证据来自2011年徐国良教授和张毅教授实验室分别背靠背发表于Science上的研究论文2,3;体内支持证据则是领域内代表性的几个实验室各自独立发现Tet蛋白介导的5mC氧化参与小鼠受精卵和原始生殖细胞(PGC)中DNA甲基化的擦除4。二是5hmC自身也可以介导特定蛋白与DNA的结合或者去结合,例如转录因子或者甲基化CpG结合蛋白等,从而参与调控基因的表达。代表性证据最早来自于Michiel Vermeulen实验室2013年发表在Cell的研究论文,通过定量质谱的蛋白质组学技术在小鼠胚胎干细胞(mESC)、神经元祖细胞(NPC)和成年小鼠脑组织中鉴定出了与5hmC结合或亲和结合5mC的特定蛋白5。
通过定量质谱以及全基因组5hmC测序,人们鉴定出了在胚胎干细胞、神经元祖细胞和成年脑组织中5hmC的水平以及基因组分布6,为研究5hmC的生物学功能迈出了关键的一步。近年来,对于DNA甲基化以及部分组蛋白修饰在早期胚胎中的分布与作用已有连续报道7,使得人们对于早期胚胎发育过程中的关键事件如合子基因组激活和全能性的建立获得了新的认知。然而,关于5hmC在胚胎发育中的功能,过去的研究手段局限于细胞免疫荧光染色,缺乏对该修饰在基因组中分布的细节刻画,使得对于胚胎中5hmC的产生以及作用仍存在较大争议。备受关注的争论之一是受精卵中产生的5hmC是发生在DNA去甲基化区域,还是从头或维持DNA甲基化产生的区域(Rachel Amouroux, et al., Nat. Cell. Biol., 2016)8;其次,尽管5hmC信号在着床前胚胎中会伴随着DNA的复制而被逐步稀释,是否仍有稳定的5hmC位点存在?其在基因组上的分布与基因表达的关系如何?最后,虽然已知Tet3蛋白介导受精卵中5hmC的产生,其它影响DNA甲基化重编程和5hmC累积的因子仍知之甚少。这些问题不仅限制了人们对5hmC在发育过程中功能机制的研究,也影响着DNA甲基化重编程如何发生——这一表观遗传学领域根本问题的结论。
2022年12月20日,中国科学院动物研究所郭帆和王红梅团队合作(中国科学院动物研究所博士生燕蕊、联合培养研究生程馨、博士生徐艳红、博士生龙鑫和北京昌平实验室副研究员古婵为本文共同第一作者),在Nature Genetics发表了题为Dynamics of DNA hydroxymethylation and methylation during mouse embryonic and germline development的研究论文。研究者对小鼠配子、受精后的着床前胚胎、着床后胚胎和原始生殖细胞以及体外培养胚胎进行了全基因组5hmC的测序分析,通过分离受精卵中的雌雄原核,定量解析了Tet3和DNA复制对于5hmC产生的影响。此外,研究者通过追踪父母本基因组,进一步揭示了父本和母本基因组之间在5hmC的产生以及分布上的异同。更进一步,研究者还发现了一个影响早期胚胎DNA甲基化重编程和5hmC累积的新因子,并利用敲除小鼠模型结合化学小分子抑制剂进行了深入解析,为胚胎和生殖细胞发育过程中5hmC的调控和功能提供了新的机制见解。
研究者发现在早期胚胎及生殖细胞发育过程中会经历两次全基因组范围5hmC的产生(图1),第一次发生于受精卵中,偏好性的累积在父本基因组上,并持续到二细胞期而在随后的卵裂过程中被逐渐稀释,直至囊胚期到达最低点;第二次发生于着床后E6.5天胚胎的上胚层细胞(epiblast)中,对称产生在父母本基因组中,在E9.5天的PGC中仍维持较高水平,而在随后阶段逐渐降低直至E13.5天达至最低点。其中,受精卵中产生的5hmC会富集在发生DNA去甲基化的位点,而不是维持性DNA甲基化或从头甲基化位点,这为5hmC可作为去甲基化的中间物提供了最为直接的体内证据。着床后胚胎中产生的5hmC则会定位于发生从头DNA甲基化的位点,高度富集于增强子(enhancer)中,在PGC发生DNA去甲基化时逐渐减少。虽然Tet3主导了受精卵中5hmC的产生,DNA复制也会影响5hmC的生成,尤其是母本基因组受DNA复制的影响更高。生殖细胞中建立的DNA甲基化印记位点会在着床前胚胎中继续维持,而在PGC阶段发生甲基化擦除。研究者发现这些印记位点在epiblast或PGC中会有较高水平的5hmC,令人意外的是在受精卵中印记位点上也有显著水平的5hmC产生。受精后产生的5hmC是否存在可以稳定维持的位点?研究者发现绝大部分位点会伴随着胚胎发育进程而逐渐丢失5hmC,然而仍有极少数位点可以维持显著水平的5hmC直至囊胚期;这些位点对应的基因在母本基因组中从卵细胞开始一直表达,而在父本基因组中则是从二细胞起始表达。
5hmC除了可以介导DNA去甲基化,是否还有其它作用?研究者发现受精后父本基因组产生的5hmC区域与合子基因组激活基因高度重合,分布于基因的启动子(promoter)、增强子和基因体(gene body)中。特定转录因子结合位点也显示出5hmC的富集(图2),Zfp57、Arntl和Ehf结合位点在卵母细胞和着床前胚胎的父母本基因组中有5hmC的富集。其中,Zfp57已被报道在小鼠早期胚胎中将DNMTs招募到其目标位点,参与印记基因甲基化的维持。Runx2, Nfil3, Thrb, Ebf1和Atf1/2结合位点在着床前胚胎的父本基因组中显著富集5hmC。参与小鼠原肠胚形成或早期胚胎器官发生的转录因子如Otx2、T、Pou5f1、Nanog、Sox2、Zic2/3和Myc在E6.5天胚胎的epiblast中也显示出其结合位点富含5hmC。生殖细胞特异的转录因子如Prdm14、Zfx、Zbtb33、Prdm9和Tcf21,其结合位点在PGC中也高度富集5hmC,这些因子已被报道在生殖细胞发育或减数分裂中发挥重要作用。进一步地,研究者分析了5hmC与特定组蛋白修饰之间的关联(图2)。受精之后,H3K4me3和H3K36me3修饰区域会富集5hmC,而H3K9me3和H3K27me3修饰位点通常会排斥5hmC的产生。卵母细胞中H3K9me2修饰区域通常与DNA高甲基化区域在空间上分离,并且从免疫荧光染色的结果推断,受精后母本基因组中H3K9me2修饰会抵抗5hmC的产生。研究者发现卵母细胞中H3K9me2修饰区域在受精后的确会排斥5hmC的产生,但仍有极少数位点会含有较高水平的5hmC。
小鼠体外培养胚胎(IVC embryos)是研究胚胎着床后形态发生和分子事件的替代模型9,其基因组中5hmC的产生与分布跟体内发育胚胎相比有何差异?研究者发现IVC胚胎总体上可以正常发生从头DNA甲基化以及基因组范围5hmC的生成,然而在一些高度富集5hmC且与胚胎形态发生和细胞分化相关联的基因位点,IVC胚胎则显示出5hmC产生不足并有着更高水平的DNA甲基化,相关基因的表达也会显著降低(图3)。这些结果表明,5hmC参与调控了E6.5天epiblast中相应基因的表达,并且IVC胚胎中特定基因位点5hmC水平降低可能与其体外发育受限相关。
除了Tet蛋白,还有哪些因子可能会影响早期胚胎中5hmC的产生和累积?在人类中,多位点印记基因疾病(MLID)与NLRP家族基因的母源突变以及DNA甲基化异常相关10。大多数人类NLRP基因在小鼠中也具有同源基因,并且Nlrp基因敲除的雌鼠通常显示出受精后着床前胚胎发育阻滞,导致不育或生育力降低。有趣的是,Nlrp KO雄鼠生育力则不受影响。研究者根据这些现象提出了一个问题,即母源缺失Nlrp是否会影响早期胚胎的DNA甲基化重编程。然而,关于NLRP基因与异常DNA甲基化之间关联的机制却鲜有文献报道,特别是在通过利用母源缺失Nlrp的小鼠来追踪异常DNA甲基化的起源方面。研究者首先系统分析了小鼠从卵细胞至早期胚胎中所有Nlrp基因的表达,发现除了已被报道的Nlrp5外,Nlrp14也高表达于卵细胞中并呈现出明显的母源表达模式。利用Nlrp14 KO的小鼠模型,研究者进行了更加深入的分析(图4)。同Nlrp5 KO小鼠类似,Nlrp14 KO雄鼠可正常生育,而KO雌鼠受精后表现为着床前胚胎发育阻滞,导致雌性不育。更进一步,研究者发现母源缺失Nlrp14的受精卵中维持性DNA甲基化酶Dnmt1及其辅因子Uhrf1会由细胞质定位转变为细胞核定位,阻碍了DNA复制关联的DNA被动去甲基化的发生,导致父本基因组DNA甲基化水平显著高于正常胚胎。此外,Nlrp14母源KO的胚胎还表现出全基因组染色质开放程度显著降低以及合子基因组激活受阻。受精卵中因Nlrp14缺失使得Dnmt1的活性增强,对5hmC的产生会有怎样的影响?研究者发现Nlrp14缺失的早期胚胎中5hmC水平显著降低,尤其是在原本会发生DNA去甲基化的区域。这一实验证据也再次表明Tet3介导的5hmC产生与DNA去甲基化紧密关联,而不是偏好性地发生在维持性DNA甲基化区域。
最后,研究者继续探索了Nlrp14是否会影响PGC中DNA甲基化重编程以及配子中DNA甲基化的建立。研究者发现Nlrp14 KO的胚胎中,PGC的发育正常且相关特征基因的表达也不受影响。同时,Nlrp14 KO的PGC中全基因组DNA甲基化的擦除亦无异常。在Nlrp14 KO后,精子和卵细胞也可以正常建立DNA甲基化,并且精子和卵子发生均无明显异常。这些结果共同表明Nlrp特异性地参与了受精卵中DNA甲基化重编程并影响Tet3介导的5hmC产生。
综上,该研究系统性地揭示了从小鼠早期胚胎发育至生殖细胞发生过程中5hmC的全基因组分布与分子调控特征,提出了早期胚胎DNA甲基化重编程的新模式(图5)。该研究不仅为表观遗传领域长期以来的重大争议问题提供了有力的实验论证,也为深入理解早期胚胎发育过程中的表观遗传调控开启了新维度。
文章链接:https://www.nature.com/articles/s41588-022-01258-x
参考文献
1. Tahiliani, M. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930-5 (2009).
2. Ito, S. et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300-3 (2011).
3. He, Y.F. et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303-7 (2011).
4. Wu, X. & Zhang, Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 18, 517-534 (2017).
5. Spruijt, C.G. et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 152, 1146-59 (2013).
6. Wu, H. & Zhang, Y. Charting oxidized methylcytosines at base resolution. Nat Struct Mol Biol 22, 656-61 (2015).
7. Du, Z., Zhang, K. & Xie, W. Epigenetic Reprogramming in Early Animal Development. Cold Spring Harb Perspect Biol 14(2022).
8. Amouroux, R. et al. De novo DNA methylation drives 5hmC accumulation in mouse zygotes. Nat Cell Biol 18, 225-233 (2016).
9. Shahbazi, M.N., Siggia, E.D. & Zernicka-Goetz, M. Self-organization of stem cells into embryos: A window on early mammalian development. Science 364, 948-951 (2019).
10. Begemann, M. et al. Maternal variants in NLRP and other maternal effect proteins are associated with multilocus imprinting disturbance in offspring. J Med Genet 55, 497-504 (2018).

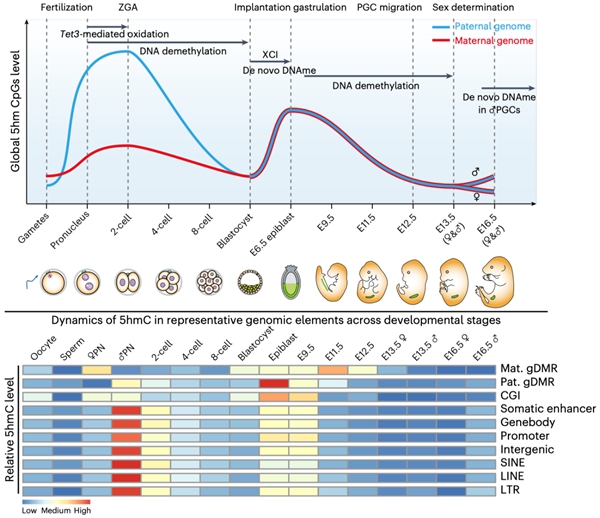
图1. 小鼠早期胚胎及生殖细胞中5hmC概观
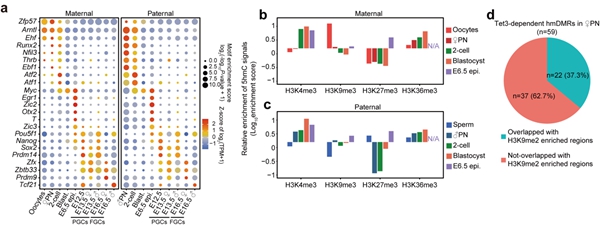
图2. 特定转录因子和组蛋白修饰与5hmC的关联
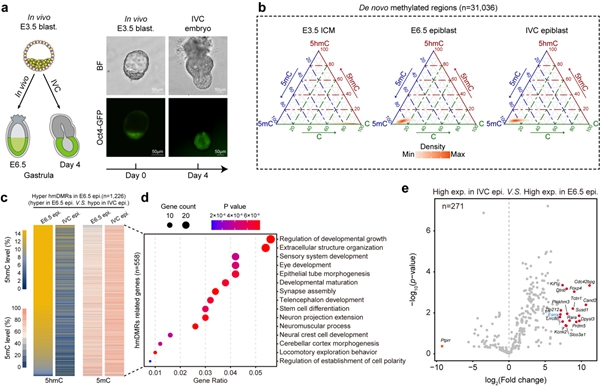
图3. IVC胚胎与体内发育胚胎的5hmC水平比较
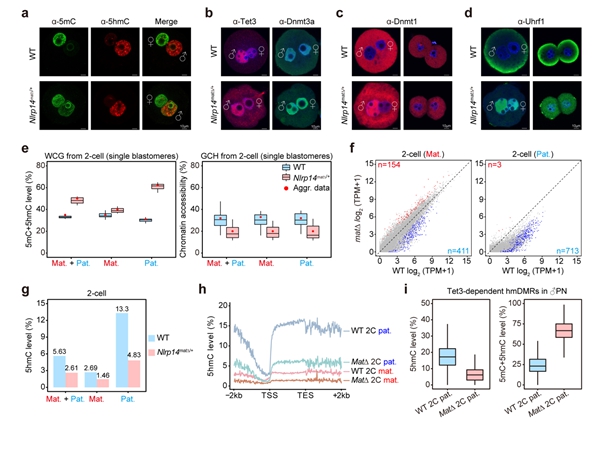
图4. Nlrp14参与早期胚胎DNA甲基化重编程并影响5hmC的累积
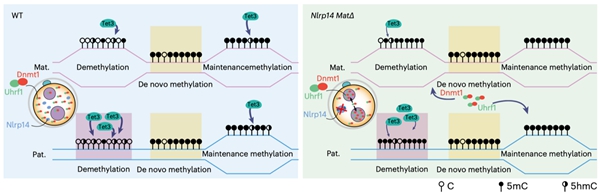
图5. 早期胚胎中5hmC产生以及DNA甲基化重编程模式
人类活动和气候变化加速生物多样性的减少,导致物种范围的转移、收缩和扩张。在全球范围内,人类活动和气候变化已对生物多样性构成了严重威胁,目前已导致全球522种灵长类动物中约68%的物种面临灭绝风险。

植物病毒素有“植物顽疾”之称,每年引起全球作物经济损失高达4000亿元。水稻作为人类重要的粮食作物之一,供给全世界一半以上的人口,主要种植于亚洲、非洲和南美洲等地区。






